Valorisation
du
dioxyde de carbone, concours général SPCL 2024.
En
poursuivant votre navigation sur ce site, vous acceptez l’utilisation
de Cookies vous proposant des publicités adaptées à vos centres
d’intérêts.
.
|
.
.
|
A.1 Etude d’un capteur de dioxyde de carbone
Pour
mesurer avec précision le taux de CO2
dans l’air, un des instruments les plus utilisés est un capteur NDIR («
Non Dispersive Infra Red »). C’est cette technologie de capteur qui est
utilisée sur l’installation pilote. L’objectif de cette partie est
d’étudier le principe de fonctionnement de ce capteur et de déterminer
le résultat d’une mesure et la résolution du capteur à partir des
acquisitions de signaux réalisés en laboratoire.
L’air circule dans une chambre de mesure en aluminium poli pour limiter
l’absorption des rayonnements par les parois. Une partie du rayonnement
émis par la source infrarouge est absorbée par les gaz. Le rayonnement
émis par la source infrarouge qui traverse le gaz est mesuré à la
sortie de la chambre de mesure par un détecteur.
La source infrarouge est une lampe à incandescence contenant un
filament de tungstène chauffé par effet Joule à une température
d’environ 3 000 K. Elle émet un rayonnement avec un large spectre dont
le maximum d’émission se situe à une longueur d’onde dépendant de la
température selon la loi de Wien : l T = 2,9 × 10−3
m‧K avec l, la
longueur d’onde en m, et T, la température en K.
1. Déterminer la
longueur d’onde du maximum d’émission de la source et vérifier qu’il se
situe dans l’infrarouge.
l=2,9
10-3 / 3000=9,7 10-7 m = 0,97 µm..
Domaine IR : [0,7 µm ; 100 µm].
Le détecteur comporte :
- une
thermopile 1 munie d’un filtre à bande très étroite, centrée autour de
4,26 μm, donnant après amplification le signal de mesure, correspondant
à une tension Um ;
- une thermopile 2 munie d’un filtre centré autour de 3,91 μm, donnant
après amplification un signalde référence, correspondant à une tension
Ur.
Une thermopile est un composant complexe fournissant une tension
proportionnelle à la température du détecteur, chauffé par le
rayonnement reçu.
La Figure 3 ci-dessous donne le spectre d’absorbance de quelques gaz
pouvant se trouver en sortie d’une cheminée.
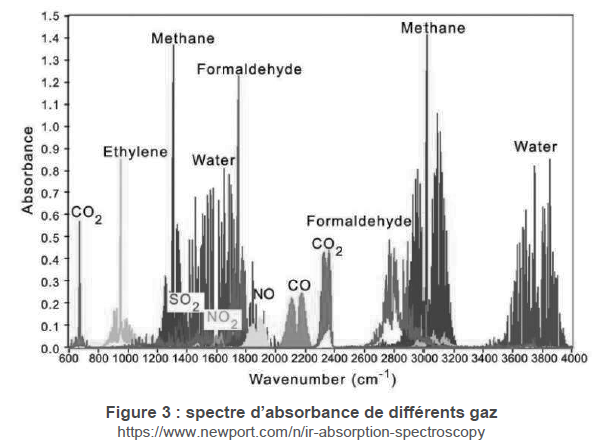
2. Justifier à
partir de la Figure 3 que le rayonnement émis par la source autour
d’une longueur d’onde de 3,91 μm est intégralement reçu par la
thermopile 2. Expliquer en quoi le signal Ur est alors une référence.
1/(3,91 10-6)=2,56 105 m-1 = 2,56 103
cm-1, valeur centrale du spectre ci-dessus.
3. Vérifier que le
filtre de la thermopile 1 est adapté à la mesure du dioxyde de carbone.
1/(4,26 10-6)=2,35
105 m-1 = 2,35 103 cm-1,
valeur correspondant au signal du CO2 du spectre ci-dessus.
4. Expliquer comment évolue le
signal Um lorsque la concentration en CO2 augmente.
L'absorbance étant proportionnelle à la concentration en CO2,
Um croît si la concentration en CO2
augmente.
Afin de
compenser la dérive due au vieillissement différent des deux
thermopiles, la source infrarouge est alimentée par une tension
alternative. Lors d’un essai en laboratoire, les signaux alternatifs
ont été obtenus, um(t ) et ur(t ), de même
fréquence que la source, reproduits sur la Figure 4 suivante :
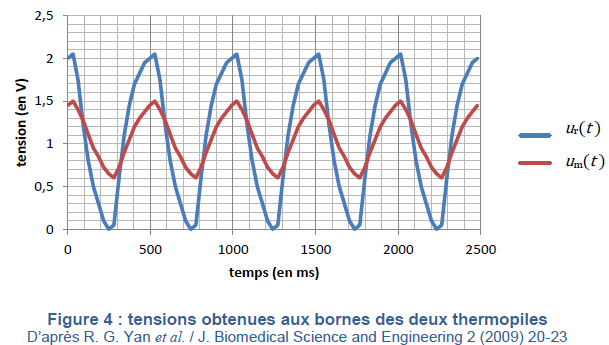
Le conditionneur de signal mesure les tensions crête à crête Um et Ur
(entre minimum et maximum) de chaque signal um(t ) et ur(t
) et les convertit en données numériques par un convertisseur
analogique-numérique à 10 bits pour une tension pleine échelle de 3,00
V. La concentration en CO2 est calculée ensuite par la
relation suivante :
c=4,04 ln (Ur/𝑈m)
5. Déterminer la
fréquence de la tension alimentant la source, ainsi que les tensions
crête à crête des signaux des deux thermopiles.
Période T = 500 ms = 0,5 s ; fréquence f = 1 / T = 1/0,5 = 2 Hz.
Tension crête à crête : Um = 0,9 V ; Ur = 2 V
6. En déduire la
concentration correspondant à cette mesure.
c = 4,04 ln(2 / 0,9)=3,2 %.
On étudie maintenant l’évolution de la résolution du capteur
(c’est-à-dire la faculté du capteur à distinguer deux valeurs de
concentration proches) en fonction de la valeur de la mesure.
7. Déterminer le
quantum de tension correspondant au convertisseur analogique-numérique.
quantum = Vréf / 210 =2 / 210=1 10-10
V.
8. Déterminer la
différence de concentration obtenue si la tension Um augmente d’un
quantum par rapport à la valeur initiale de 0,850 V.
c1 =4,04 ln(2 /(0,85 +0,85 10-10)=3,46 %.
3,46-3,2 = 0,26 %.
9. Déterminer
la différence de concentration obtenue si la tension Um augmente d’un
quantum par rapport à une valeur initiale de 0,020 V.
c1 =4,04 ln(2
/(0,02 +0,85 10-10)~ -2 10-8 % ~0
La différence de concentration est nulle.
10. Conclure sur l’évolution de la
résolution en concentration du capteur lorsque la concentration
augmente.
La résolution est bonne si la concentration en dioxyde de carbone n'est
pas trop faible.
A.2 Etude de la capture du
dioxyde de carbone
Afin de capturer le dioxyde de carbone émis par les centrales
thermiques, les gaz sortant des cheminées passent dans une colonne à
garnissage dans laquelle circule à contre-courant une solution aqueuse
d’hydroxyde de sodium, schématisée sur la Figure 5 ci-dessous.

Le gaz sortant des cheminées, composé d’air et de dioxyde de carbone,
entre dans la colonne par le bas (pied de colonne). La solution aqueuse
d’hydroxyde de sodium est envoyée par le haut (tête de colonne). Une
partie du dioxyde de carbone se dissout dans le liquide. Il sort alors
de la colonne un gaz constitué d’air épuré par en haut et une solution
aqueuse chargée en CO2 par en bas. L’efficacité de la
capture est d’autant plus forte que :
- la fraction molaire de CO2 dans le gaz sortant est faible ;
- la quantité de CO2 dissous dans le liquide est grande.
Caractéristiques de l’installation :
- débit de liquide réglable de 10 à 100 % par une pompe (débit maximal
de 16 L‧h-1) ;
- débit de liquide réglé à L = 50 % ;
- débitmètre de gaz entrant gradué en % (100 % correspond à 80 L‧min-1)
;
- débit de gaz G réglé à 45 % ;
- température du liquide à l’entrée 18 °C ;
- hauteur de la colonne 1,9 m ;
- pression en tête de colonne égale à la pression atmosphérique (1,01
bar).
11. Sachant que la
solubilité des gaz diminue lorsque la température augmente, justifier
que le liquide doit être froid pour améliorer l’efficacité de la
capture.
Le gaz est plus soluble dans un liquide froid que dans un liquide
chaud. L'efficacité de la colonne augmente si la température du liquide
diminue.
Pour que le mélange entre les phases gazeuse et liquide soit efficace,
il faut maintenir une différence de pression approximative entre le
pied et la tête de colonne de 8 mbar par mètre de colonne. L’abaque de
la Figure 6 ci-dessous représente la variation de la pression relative
en pied de colonne (différence de pression entre le gaz en pied de
colonne et l’air extérieur) en fonction du
débit de gaz circulant, pour plusieurs débits de liquide.
8x1,9=15,2
mbar.
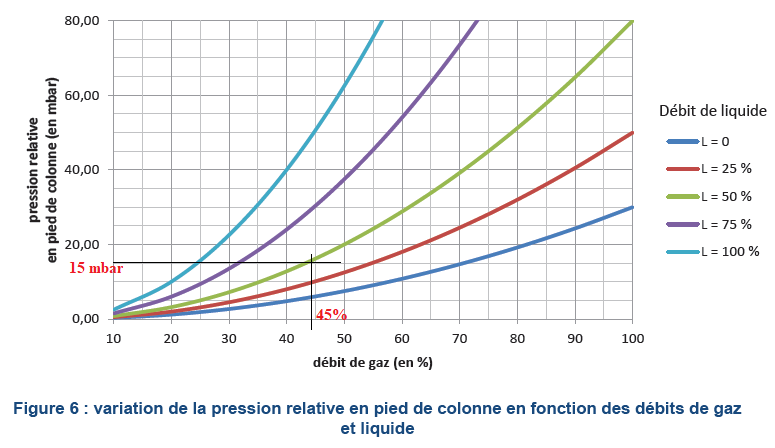
12. Vérifier que
les réglages choisis conduisent à un fonctionnement correct de la
colonne.
- débit de liquide réglable de 10 à 100 % par une pompe (débit maximal
de 16 L‧h-1) ;
- débit de liquide réglé à L = 50 % soit 8 L h-1.
- débitmètre de gaz entrant gradué en % (100 % correspond à 80 L‧min-1)
;
- débit de gaz G réglé à 45 % soit 80 x0,45=36 L min-1.
Afin d’empêcher la
sortie du gaz par le bas, il est nécessaire de maintenir un niveau de
liquide constant en pied de colonne. Pour cela, le liquide sort par un
tube assez long (garde liquide) qui remonte jusqu’à une hauteur h
réglable, comme précisé sur le schéma ci-dessous, représentant la
partie inférieure de la figure 5.
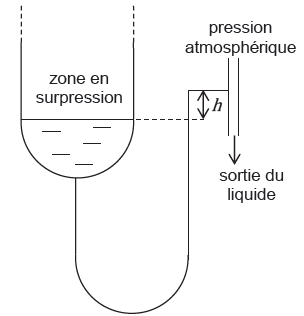
13. Déterminer la
hauteur h minimale pour faire tout juste couler le liquide si le pied
de colonne est en surpression de 16 mbar par rapport à la pression
atmosphérique (on considérera la vitesse suffisamment petite pour
pouvoir appliquer le principe fondamental de la statique des fluides
incompressibles).
16 mbar soit 16 x 100 = 1,600 Pa.
1600 = r g h =
1000 x9,81 h ; h =0,16 m.
|
Les
réglages de débits de gaz effectués correspondent à une quantité de
matière totale de gaz entrantde 1,51 mole par minute. La fraction
molaire de CO2 dans ce gaz entrant, mesurée par un capteur
du type étudié dans la partie A.1, vaut 12,0 %.
14. Calculer la quantité de matière de dioxyde de carbone entrant dans
la colonne en une minute.
1,51x0,12=0,18 mol / minute.
Le CO2 réagit avec l’hydroxyde de sodium en excès pour
donner du carbonate de sodium (2 Na+(aq)+ CO32-(aq)).
15. Écrire
l'équation de la réaction modélisant la transformation prenant place
entre le dioxyde de carbone et l'hydroxyde de sodium.
CO2(aq) +2HO-aq
--> CO32-aq + H2O(l).
On
recueille 1,42 L de solution aqueuse sortant pendant 10 min. On prélève
5,0 mL de cette solution qu’on titre par de l’acide chlorhydrique
(monoacide fort) de concentration 0,87 mol‧L-1. Le graphe
suivant (Figure 7), représentant le pH et sa dérivée en fonction du
volume d’acide versé, est obtenu. La première équivalence correspond à
la neutralisation des ions hydroxyde restant, les deux suivantes aux
ions carbonate (dibase).
16. Écrire
l’équation de la réaction modélisant la transformation prenant place
entre l’ion carbonate et l’acide chlorhydrique.
CO32-aq +2H3O+aq
--> CO2aq + 3H2O(l).
17. Déterminer la quantité de
matière d’ion carbonate présent dans l’échantillon prélevé.
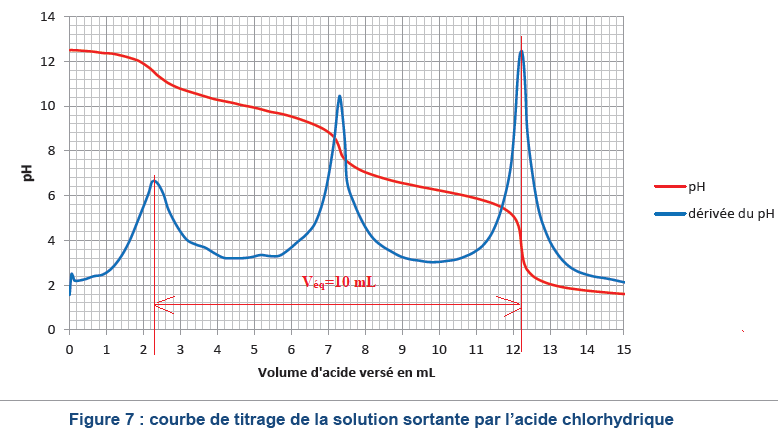
Quantité de matière d'acide chlorhydrique : 10 x 0,87 = 8,7 mmol.
Quantité de matière d'ion carbonate 0,5 x8,7 = 4,35 mmol dans 5,0 mL de
solution.
18. En déduire la
quantité de carbonate de sodium dans la solution prélevée pendant 10
min.
4,35 x1,42 103 / 5=1,2354 103 ~1,24 103
mmol =1,24 moles en 10 minutes ou 0,124 mol en une minute.
Celle-ci correspond à la quantité de matière de CO2 dissous
pendant cette même durée.
En utilisant de l’eau pure à la place de la solution d’hydroxyde de
sodium, avec les mêmes réglages, on trouve un débit en quantité de
matière de CO2 dissous dans l’eau de 0,007 mol‧min-1.
19. Conclure sur
l’intérêt d’utiliser une solution aqueuse d’hydroxyde de sodium au lieu
d’eau pure pour la capture du dioxyde de carbone.
0,124
/ 0 007 ~18.
Une
solution aqueuse d’hydroxyde de sodium permet de capturer 18 fois plus
de dioxyde de carbone que l’eau pure.
B. Valorisation du dioxyde
de carbone.
Une alternative au stockage du dioxyde de carbone après son
captage est son utilisation dans des procédés permettant de le
valoriser, c’est-à-dire d’utiliser cette matière première disponible en
grandes quantités et de la convertir en produits à plus forte valeur
ajoutée. Actuellement, environ 230 Mt/an de dioxyde de carbone sont
engagées dans ce processus CCU (Carbon Capture and Utilisation)1. Le
dioxyde de carbone peut être utilisé directement en tant que fluide
réfrigérant, solvant, ou encore gazéifiant de boissons, être transformé
en carburants ou molécules organiques d’intérêt (valorisation chimique)
ou encore servir dans des processus de méthanation biologique ou
fermentation biologique (valorisation biologique). Cette partie
s’intéresse à quelques applications industrielles parmi les différentes
voies citées.
B.1 Valorisation chimique
en acide salicylique.
Il existe diverses voies chimiques de valorisation du dioxyde de
carbone, la principale en termes de volume étant la synthèse d’urée.
Nous nous limiterons dans cette partie à quelques exemples
d’utilisation du CO2 comme précurseur de molécules
d’intérêt. Le CO2
est utilisé industriellement pour former de l’acide salicylique.
L’acide salicylique est utilisé directement dans certains produits
dermatologiques ou cosmétiques en raison de ses propriétés
antiseptiques et antalgiques. Il est également un précurseur de l’acide
acétylsalicylique (ou aspirine).
B.1.1 Production de
l’acide salicylique.
Cette partie s’attache à étudier les mécanismes mis en jeu dans la
production de l’acide salicylique et à quantifier la masse de CO2
valorisée par cette filière.
20. Identifier les
fonctions chimiques présentes dans l’acide salicylique.
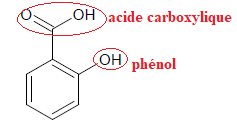
L’acide salicylique est obtenu industriellement par carboxylation du
phénol, cette transformation chimique est modélisée par la réaction de
Kolbe-Schmitt dont l’équation est donnée.
Un mécanisme simplifié de cette réaction est proposé ci-dessous.
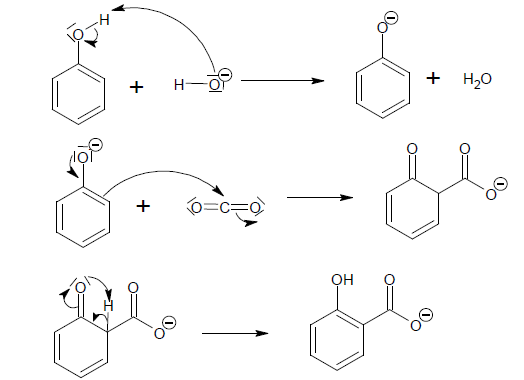
21. Expliquer la
première étape de ce mécanisme.
Réaction acide base entre la base forte HO- et l'acide
faible C6H5OH.
22. Indiquer le
type de réaction mise en jeu lors de la deuxième étape.
Substitution électrophile sur le noyau benzénique.
23. Recopier et
compléter la dernière étape en ajoutant la(les) flèche(s) courbe(s)
adéquate(s).
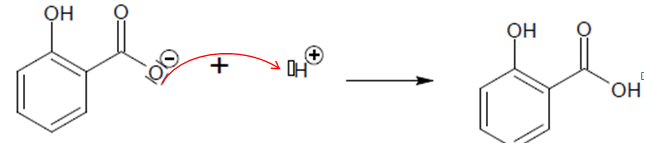
La production mondiale annuelle d’acide salicylique est de 70 000 t/an.
(7 1010 g / an)
24. Estimer la masse de CO2
annuelle valorisée selon cette filière.
Masse molaire acide salicylique : 138 g / mol.
Quantité de matière d'acide salicylique : 7 1010 /138=5,1 108
mol.
Masse de dioxyde de carbone : 5,1 108 xM(CO2 )= 5,1 108 x44 =2,2 1010
g ou 2,2 104 tonnes.
B.1.2 Acide salicylique, composant d’un
antalgique.
Outre son utilisation comme précurseur de l’aspirine, l’acide
salicylique est utilisé en tant que teldans certains produits
dermatologiques ou cosmétiques en raison de ses propriétés
antiseptiques et antalgiques. Il entre notamment dans la composition du
Synthol®, un antalgique d’action locale utilisé en cas d’ecchymoses ou
contusions.
Lors d’une séance de travaux pratiques en laboratoire, un groupe
d’étudiants cherche à vérifier si la teneur en acide salicylique d’un
échantillon de Synthol® est conforme à l’étiquetage. Les étudiants
mettent en oeuvre un dosage spectroscopique par étalonnage. Des mesures
classiques d’absorbance ne peuvent pas être utilisées, car d’autres
espèces présentes dans le Synthol® absorbent la lumière UV à la même
longueur d’onde que l’acide salicylique. En revanche, après absorption
de la lumière, l’acide salicylique réémet une partie de cette énergie
absorbée sous forme de rayonnement avec une longueur d’onde plus élevée
: c’est le phénomène de fluorescence. L’intensité de la lumière émise,
appelée intensité de fluorescence et notée IF, s’écrit sous
la forme IF=kc où k est une constante et c la concentration
en quantité de matière de l’acide salicylique dans l’échantillon. Cela
permet d’exploiter cette intensité de fluorescence pour mettre en
oeuvre un dosage par étalonnage.
Choix de la longueur
d’onde d’excitation.
Lors de la mise en oeuvre du dosage, l’échantillon est soumis à un
rayonnement de longueur d’onde égale à 295 nm, qui est absorbé par
l’acide salicylique ; cette phase est appelée excitation. Le spectre
d’absorption d’une solution d’acide salicylique obtenu en laboratoire
est donné ci-dessous.
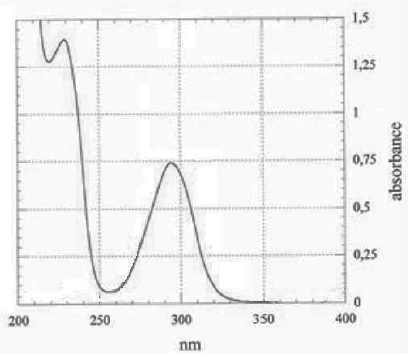
25. À partir du
spectre d’absorption, justifier le choix de la longueur d’onde
d’excitation.
Pour une meilleure précision, on se place au maximum d'absorption soit
295 nm.
Un diagramme, simplifié, représentant quelques niveaux d’énergie de la
molécule d’acide salicylique permet de comprendre les phénomènes mis en
jeu :
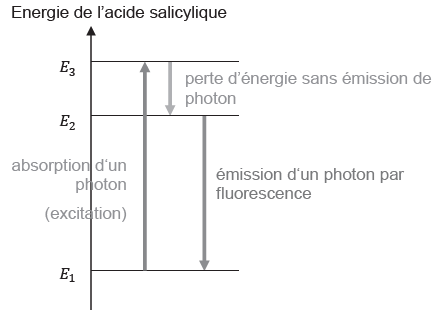
26. À partir du
diagramme d’énergie, expliquer pourquoi la longueur d’onde émise est
supérieure à la longueur d’onde absorbée lors de l’excitation.
Longueur d'onde absorbée = h c / (E3-E1).
Longueur d'onde émise =h c /(E2-E1).
E2-E1 <
E3-E1,
donc la longueur d'onde émise est supérieure à la longueur d'onde
absorbée.
Réalisation du dosage par
les étudiants.
Les étudiants mettent en oeuvre le dosage par étalonnage pour
déterminer la teneur en acide salicylique dans un échantillon de
Synthol® et vérifier la conformité avec l’étiquette.
Le protocole de la manipulation est donné ci-après.
- On prépare 50 mL d’une solution-mère d’acide salicylique dans l’eau
distillée, à la concentration c0 = 1,0.10-2 mol·L-1,
en présence de 1 mL d’éthanol (pour aider à la
dissolution).
- À partir de cette solution-mère, on prépare une solution F0
à la concentration c’0 = 2,0∙10-4 mol·L-1,
par dilution dans une solution-tampon à pH = 7,0.
- On prépare ensuite une série de cinq solutions-filles F1 à
F5 de concentration 2,0.10-6 à 10,0.10-6
mol·L-1, en diluant la solution F0.
27. Proposer un
protocole pour préparer la solution F0 à partir de la
solution-mère. Le matériel utilisé sera précisé.
Facteur de dilution F = c0 /c’0 =50.
Dans une fiole jaugée de 1,0 L contenant 1 /3 d'eau distillée ajouter
1000 / 50 = 20,0 mL de solution mère à l'aide d'une pipette jaugée.
Agiter. Compléter avec de l'eau distillée jusqu'au trait de jauge.
Agiter pour rendre homogène.
Les résultats de mesure
d’intensité de fluorescence émise pour les solutions-filles, ainsi que
la courbe d’étalonnage associée sont donnés ci-dessous :
Solution
|
F1
|
F2
|
F3
|
F4
|
F5
|
concentration
(mol/L)
|
2,0
10-6
|
4,0
10-6 |
6,0
10-6 |
8,0
10-6 |
10,0
10-6 |
IF
|
13,1
|
25,5
|
36,4
|
49,9
|
63,5
|
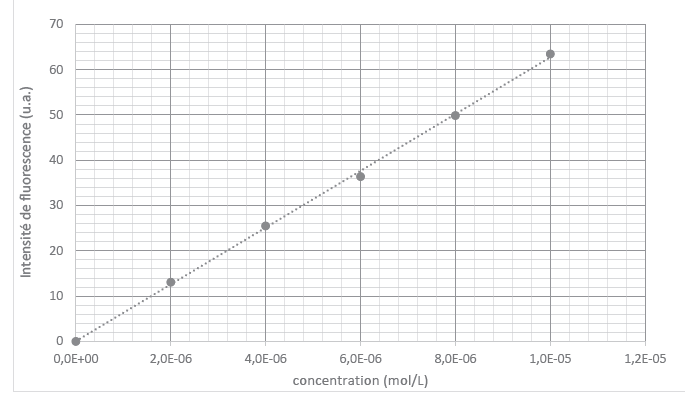
28. Commenter
l’allure de la courbe d’étalonnage.
IF est proportionnelle à la concentration.
29. À partir
de cette courbe, déterminer la valeur de la constante k définie en
début de cette sous-partie
k = coefficient directeur de la droite =50 / (8,0 10-6)
=6,25 106 L mol-1.
B.1.2. Chacun des
15 étudiants a ensuite mesuré l’intensité de fluorescence d’une
solution de Synthol® dilué 100 fois.
Donnée : densité du Synthol, d = 0,93.
Expliquer pourquoi il a été nécessaire de
diluer le Synthol®. La réponse devra être justifiée par un calcul.
Acide salycilique 0,0105 g pour
100 g de solution.
Volume de 100 g de solution : 100 / 0,93 = 107,5 mL.
M(acide salycilique) = 138 g/mol.
Quantité de matière en acide salycilique : 0,0105 / 138 = 7,6 10-5
mol.
Concentration en acide salycilique : 7,6 10-5 / 0,1075 =7,1
10-4 mol/ L valeur très supérieure aux concentrations de la
gamme d'étalonnage.
Il faut donc diluer environ 100 fois le Synthol.
31.
Un des étudiants a mesuré une intensité de fluorescence IF
égale à 45,1 u.a. Déterminer la masse d’acide salicylique pour 100 g de
Synthol® correspondant à cette mesure.
c = IF / k =45,1 /(6,25 106) = 7,216 10-6
mol / L.
7,216 10-6 x 138 =9,96 10-4 g / L.
Masse de 1 L =930 g.
Masse d'acide salycilique dans 100 g de synthol : 9,96 10-4
x100 / 930=1,07 10-4 g.
Tenir compte de la dilution :1,07 10-4 x100=0,0107 g.
La prise en compte des résultats des 15 étudiants conduit à une valeur
moyenne de 0,01073 g d’acide salicylique pour 100 g de Synthol®, avec
un écart-type de 0,0005 g. On rappelle que l’incertitude-type sur la
moyenne d’une série de valeurs est égale à l’écart-type de cette série
divisée par N½ où N est le nombre de mesures.
32. Exprimer le
résultat de mesure moyen des 15 étudiants avec le nombre de chiffres
significatifs adaptés et l’incertitude-type associée.
Incertitude type = 0,0005 / 15½ =1,3 10-4 g.
0,0107 ±2 10-4 g.
33. Evaluer la
différence entre le résultat obtenu expérimentalement et la valeur de
référence de l’emballage en nombre d’incertitudes-types. Conclure.
Acide salycilique 0,0105 g pour
100 g de solution.
Différence : 0,0107 -0,0105 =2 10-4 soit une
incertitude-type.
B.2 Valorisation en
polycarbonate.
Outre la production d’urée et d’acides carboxyliques comme l’acide
salicylique, le CO2 est utilisé dans l’industrie chimique
pour produire des polycarbonates qui sont des composés conduisant à de
nombreuses applications du fait de leurs propriétés thermiques,
optiques et mécaniques. En particulier, le polycarbonate de bisphénol A
(appelé plus couramment polycarbonate) possède une grande résistance
aux chocs, permettant son utilisation dans de nombreux objets du
quotidien comme des casques de moto ou des coques de smartphones.
B.2.1 Résistance aux chocs
lors d’une chute libre sans frottements
Pour être homologuées, les coques de téléphones doivent vérifier
certaines normes. En particulier, il existe une certification
MIL-STD-810G délivrée par le laboratoire de test de développement
technologique de l’armée Américaine, qui assure entre autres une
résistance à 26 chutes de 1,2 mètre.
Le polycarbonate présente une résistance aux chocs de 10 kJ·m-2.
À titre de comparaison, dans cette partie on cherche à estimer
l’énergie surfacique (exprimée en J.m-2) libérée lors d’un
choc consécutif à la chute d’un smartphone d’une hauteur de 1,20 m.
Dans le référentiel terrestre supposé galiléen, on étudie la chute d’un
smartphone de masse m = 210 g. On le laisse tomber depuis une hauteur
du sol h = 1,2 m, sans vitesse initiale. Il est dans une position
horizontale lors du lâcher. On considère dans cette première étude que
les frottements dus à l’air lors de la chute du smartphone sont
négligeables.
Donnée : intensité du champ de pesanteur : g = 9,81 m·s-2.
34. En prenant
comme référence une énergie potentielle nulle au niveau du sol,
exprimer l’énergie potentielle de pesanteur du smartphone à l’instant
initial.
Ep = mgh = 0,210 x9,81 x1,2 =2,47 J.
35. Indiquer la
valeur de l’énergie cinétique initiale du smartphone.
Ec = 0 la vitesse initiale étant nulle.
36. À l’aide d’un
bilan d’énergie mécanique, en déduire la valeur d’énergie cinétique
acquise par le téléphone au moment où il touche le sol.
Conservation de l'énergie mécanique :
Ep+Ec = 2,47 J.
L'énergie potentielle étant nulle au sol, l'énergie cinétique au sol
est de 2,47 J.
37. En proposant
des estimations raisonnables pour les valeurs des différentes surfaces
du smartphone, donner une estimation de l’énergie cinétique surfacique
(en J·m-2), dans le cas où le smartphone tombe à plat, puis
dans le cas où il est lâché en position verticale et tombe sur la
tranche.
Il tombe à plat : surface : 0,20 x0,10 = 0,02 m2.
2,47 / 0,02 =124 J m-2. (124 x26~3 kJ m-2)
Il tombe sur la tranche : surface = 0,20 x 0,02 =0,004 m2.
2,47 /0,004=617 J m-2. (617 x26~16 kJ m-2)
38. Comparer ces valeurs à la
résistance aux chocs annoncée pour le polycarbonate. Commenter.
Le smartphone résiste bien à 26 chutes s'il
tombe à plat.
B.2.2 Amélioration du
modèle de la chute.
Le modèle de la chute libre sans frottement est questionné. Dans cette
partie, on cherche à vérifier si la force de frottement f est
négligeable lors de la chute ou si elle est modélisable.
À l’aide de l’accéléromètre du smartphone, on a relevé, lors de sa
chute, les valeurs expérimentales de son accélération verticale en
fonction du temps pour une durée totale de 1 s, l’instant t = 0
correspondant au moment où le smartphone est lâché, sans vitesse
initiale depuis une hauteur du sol h = 1,20 m. Un coussin est placé au
sol pour réceptionner l’appareil. La courbe obtenue est donnée
ci-dessous.

39. À partir de
cette courbe, estimer la durée de la chute en repérant le moment de
l’impact au sol.
Durée de la chute : 0,046 s.
40. Justifier le signe de
l’accélération au moment de l’impact puis donner une explication aux
oscillations visibles en fin d’acquisition.
Accélération au moment de l'impact =( 0 - vitesse juste avant l'impact)
/ durée de cette variation de vitesse.
Le smartphone rebondit plusieurs fois avant de s'immobiliser.
On cherche à comparer cette courbe expérimentale avec trois
modèles de chute différents.
- Modèle 1. Chute libre sans frottements :f=0.
- Modèle 2. Présence d’une force de frottement fluide f modélisée en
norme par : f=kv .
- Modèle 3. Présence d’une force de frottement fluide modélisée en
norme : f=k'v2. où k et k' sont les coefficients de
frottement fluide (respectivement en N·s·m-1 et N·s2·m-2).
Le smartphone, de masse m = 210 g, est lâché sans vitesse initiale
depuis une hauteur de 1,2 m du sol. Le repère terrestre d’étude (O, x,
y) est représenté par le schéma ci-après, l’origine du repère étant
placée sur la position initiale du smartphone y(0) = 0 :
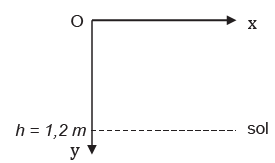
On s’intéresse d’abord au modèle 1 sans frottements.
43. Dans le cadre
de ce modèle 1, déterminer l’expression de la vitesse verticale vy(t)
du smartphone en fonction du temps, puis celle de sa position verticale
y(t) en fonction du temps.
vy(t) = gt ; y(t) = ½gt2.
44. Déterminer la
valeur de la durée de la chute et conclure si cette valeur est en
accord avec la courbe expérimentale.
1,2 = 0,5 x9,81 t2 ; t2 =0,245 ; t ~0,5 s. Accord
avec la courbe expérimentale.
45. Commenter
l’allure de l’accélération expérimentale avant l’impact en indiquant si
elle est en accord avec le modèle 1.
L'accélération diminue légèrement avant l'impact. Donc désaccord
avec le modèle 1.
On cherche maintenant à affiner le modèle en déterminant, le cas
échéant, le modèle avec frottements le plus en adéquation avec la
courbe expérimentale.
Les mesures expérimentales d’accélération ont été faites à intervalle
de temps régulier Dt.
On obtient ainsi un ensemble de points discrets, le point 0
correspondant à la mesure à t = 0, le point 1 à t1= Dt , le point 2 à t2 = 2Dt, etc., avec Dt = ti+1-ti.
46. Exprimer ai,
l’accélération au point i, en fonction de la vitesse vi au
point i, de la vitesse vi+1 au point i+1 et de l’intervalle
de temps Dt.
ai = (vi+1-vi) / Dt.
47. En déduire
l’expression de la vitesse vi+1 en fonction des
caractéristiques en i et de Dt.
vi+1-vi= ai Dt. vi+1-vi= ai Dt +vi
En
procédant par itération et en considérant que la vitesse initiale est
nulle, on peut ainsi estimer les valeurs de la vitesse pour les
différents instants de l’expérience. Le tracé de l’accélération en
fonction de la vitesse, obtenu à partir des résultats expérimentaux
précédents, est donné ci-dessous, en se limitant aux points
correspondants à la durée de la chute avant l’impact.
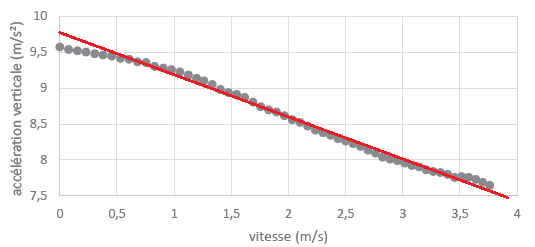
48. Commenter cette
courbe en précisant si les frottements sont négligeables durant toute
la durée de la chute. S’ils ne le sont pas, préciser le modèle semblant
le plus en adéquation avec l’expérience et estimer une valeur du
coefficient de frottement fluide correspondant.
L'acélération n'étant pas constante, il faut prendre en compte les
frottements.
La seconde loi de Newton conduit à : a = g -f / m= g- k / m v pour le
modèle 2.
a = g-k'/m v2 pour le modèle 3.
Les points sont à peu près alignés, le modèle 2 peut convenir.
Valeur absolue du coefficient directeur de la droite rouge :
k / m=(9,8-7,5) / 3,8 ~0,6 ; k =0,6 x0,21=0,13 N·s·m-1.
18 B.3 Valorisation sans transformation : utilisation comme fluide réfrigérant
Par son caractère inerte, stable et non corrosif, le dioxyde de carbone
représente une alternative à d’autres fluides réfrigérants avec un
pouvoir de réchauffement beaucoup plus important que le CO2.
Les machines réfrigérantes usuelles présentent des coefficients de
performance (COP), définis comme le rapport entre la puissance utile et
la puissance coûteuse, dont les valeurs avoisinent 3 ou 4. Le principal
inconvénient de l’utilisation du CO2 comme fluide
frigorigène est qu’il nécessite des pressions élevées (plusieurs
dizaines de bars), ce qui pose des problèmes d’étanchéité du matériel
et de sécurité. Pour pallier cette difficulté, on peut mettre en oeuvre
des systèmes en cascade à deux fluides frigorigènes, l’un étant le CO2
qui est condensé grâce à l’évaporation du deuxième fluide. On se
propose ici d’étudier quelques caractéristiques d’une machine
réfrigérante constituée d’un système en cascade CO2/NH3 et de déterminer le coefficient de performance (COP) de cette machine réfrigérante.
Description des échanges mis en jeu dans le système lors de chaque transformation.
1-2 : le CO2 reçoit une puissance mécanique PPCOmCO2 dans le compresseur ;
2-3 : le CO2 est refroidi jusqu’à la température TCondCO2 puis condensé à TCondCO2 dans l’échangeur et fournit une puissance thermique PPEchCO2 ;
3-4 : le CO2 passe dans un détendeur, sans échange thermique ni mécanique ;
4-1 : le CO2 reçoit une puissance thermique PPEvapCO2 de la part de la source froide (espace à réfrigérer) et subit une évaporation à la température TTEvCO2 puis un réchauffement ; 5-6 : le NH3 reçoit une puissance mécanique PPComp NH3 dans le compresseur ;
6-7 : le NH3 est refroidi jusqu’à la température TTCond NH3 puis condensé à la température TTCond NH3, et fournit une puissance thermique PPCond NH3 au milieu ambiant ;
7-8 : le NH3 passe dans un détendeur, sans échange thermique ni mécanique ;
8-5 : le NH3 reçoit une puissance thermique PPEch NH3 et subit une évaporation à la température TTEvap NH3 dans l’échangeur.
49. Compléter le document en indiquant dans chaque encadré la puissance échangée correspondant à chaque flèche.

50. Indiquer dans quel état physique se trouve le CO2 aux points 1, 2, 3 et 4 du circuit basse température.
1et 2 : gaz ; 3et 4 : liquide.
L’ensemble des tuyaux ainsi que l’échangeur sont supposés parfaitement
calorifugés. Toutes les pertes sont donc négligées. Le débit massique
du fluide correspondant au point i est noté DDi et le régime est stationnaire.
51. Donner une relation entre les débits massiques DD1, DD2, DD3 et DD4. De même, donner une relation entre DD5, DD6, DD7 et DD8.
Conservation du débit : DD1= DD2= DD3 = DD4.
Conservation du débit : DD5= DD6= DD7 = DD8.
On s’intéresse aux transformations se déroulant dans l’échangeur.
52. Donner la relation entre PPEch CO2 et PPEch NH3, sachant que les pertes thermiques sont négligées.
PPEch CO2 = PPEch NH3.
Le terme PPEch CO2 comprend en fait deux contributions :
- la puissance PPRefr CO2 fournie par le refroidissement du CO2 de TT2 = 60 °C à TTCond CO2 = 0 °C
- la puissance PPCond CO2 fournie par la condensation du CO2 sous une pression de 35 bar.
L’énergie thermique massique libérée par la condensation du CO2 à la température TTCond CO2 est de 247 kJ·kg-1. Par ailleurs, la capacité thermique massique du CO2 lors du refroidissement de TT2 à TTCond CO2 est C= 0,85 kJ·kg-1·°C-1. Le débit massique DD2 = 6,4.10-3 kg·s-1 est donné (source 4).
53. Déterminer la valeur de la puissance PPEch CO2.
PPRefr CO2 = C DD2 (TT2 - TTCond CO2 )=0,85 x6,4 10-3(60-0)=0,326 kW.
PPCond CO2 =247 x 6,4.10-3 =1,58 kW.
PPEch CO2= 0,326 + 1,58~1,91 kW.
Par ailleurs, l’énergie massique reçue par NH3 dans l’évaporateur est de 1090 kJ·kg-1.
54. Déterminer la valeur du débit massique D5.
1090 D5 = 1,91 ; D5 = 1,73 10-3 kg·s-1 .
55. À l’aide d’un bilan d’énergie, exprimer PPEvap CO2 en fonction de PPComp CO2 et PPEch CO2.
PPEvap CO2 =PPComp CO2 + PPEch CO2.
On rappelle que le coefficient de performance (COP) d’une machine réfrigérante est défini par l’expression suivante :
COP=puissance utile / puissance coûteuse
56. Exprimer le
coefficient de performance, COP, de la machine réfrigérante étudiée en
fonction de certaines des puissances définies.
Les puissances mécaniques reçues par les deux compresseurs ont pour valeurs :
PPComp CO2 = 0,47 kW et PPComp NH3 = 0,46 kW.
Puissance coûteuse = PPCOmCO2 +PPComp NH3 =0,47 +0,46=0,93 kW.
Puissance utile = PPRefr CO2 +PPEch NH3.=1,91+1,91= 3,82 kW.
57. En
déduire la valeur du COP de cette machine réfrigérante dans les
conditions de fonctionnement. Conclure sur l’efficacité de cette
machine réfrigérante.
COP = 3,82 / 0,93 ~4,0
|
|
|