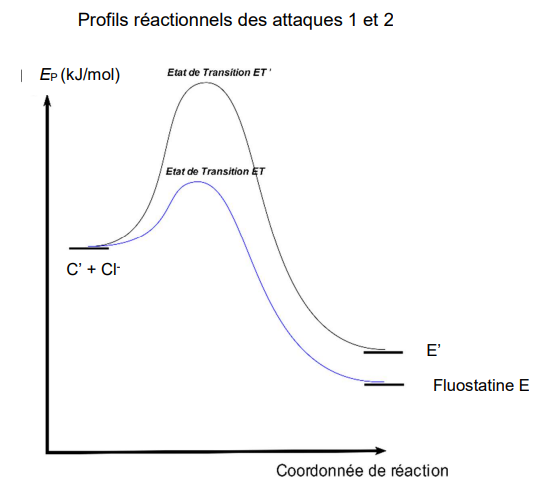
Étude structurale.
1. Identifier 5 groupes caractéristiques, présents dans les
cycles (1), des membres de la famille des fluostatines. Indiquer le nom de la famille
fonctionnelle correspondante. Indiquer également le nom d’une fluostatine contenant le groupe
caractéristique concerné.
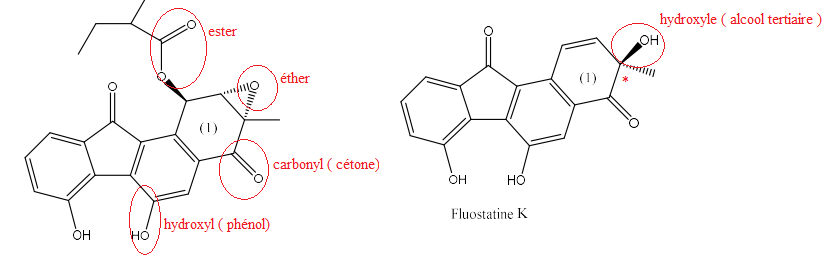 2.
2. Justifier que la fluostatine K est chirale.
Cette molécule possède un atome de carbone asymétrique ( repéré par *).
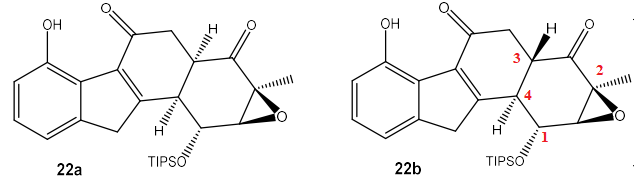
3. Repérer le(s) centre(s) stéréogène(s)
et établir, en justifiant, leur(s) descripteur(s) stéréochimique(s) de la fluostatine ( C).
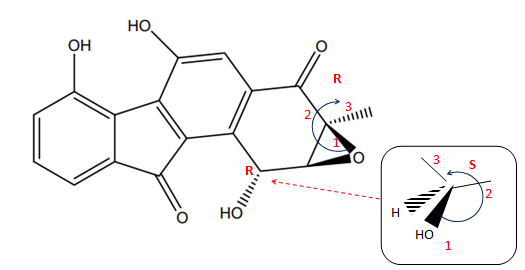 4.
4. Dénombrer, en justifiant votre réponse, le nombre de stéréoisomères associés à la fluostatine
E. Expliquer pourquoi la synthèse doit préférentiellement mener à un stéréoisomère donné.
Deux atomes de carbone asymétriques, donc 2
2 = 4 stéréoisomères.
L'activité antitumorale dépend de la structure.
Extraction d'un milieu de culture.
La fluostatine C a d’abord été obtenue en utilisant la souche de bactéries Streptomyces Acta 1383.
Cette bactérie cultivée dans des cuves de fermentations de 20 L fournit des quantités de fluostatines
détectables et quantifiables par Chromatographie Liquide à Haute Pression.
Le laboratoire de contrôle qualité s’interroge sur la possibilité de mettre au point une méthode
spectrophotométrique de dosage par étalonnage permettant de déterminer la quantité de fluostatine C
produite par ce procédé. Cette méthode doit pouvoir quantifier une concentration d’au moins 6 10
-
6 mol∙L
-1 de fluostatine C. Le spectrophotomètre UV-visible utilisé est un spectromètre Cary®
50 UVVis. La cuve spectrophotométrique utilisée, en UV-Silica, laisse passer la lumière dont la longueur
d’onde est comprise entre 230 nm et 2500 nm.
5. Déterminer la longueur d’onde de travail la plus adaptée pour détecter la fluostatine C dans les
conditions de travail décrites.
Pour une meilleure précision, on se place à une longueur d'onde pour
laquelle le spectre présente un maximum d'absorption ( par exemple 260
nm ).
On réalise une gamme d’étalonnage de cinq solutions de Fluostatine C de concentrations connues.
Leurs absorbances sont mesurées avec trois répétitions de mesure pour chaque solution étalon.
Les résultats obtenus sont décrits ci-après :
Solution étalon
|
n°1
|
n°2
|
n°3
|
n°4
|
n°5
|
Concentration en fluostatine C ( mol / L)
|
1,00 10-5
|
3,00 10-5 |
5,00 10-5 |
7,00 10-5 |
9,00 10-5 |
Absorbance essai 1
|
0,086
|
0,259
|
0,432
|
0,605
|
0,778
|
| Absorbance essai 2 |
0,083
|
0,251
|
0,439
|
0,608
|
0,783
|
| Absorbance essai 3 |
0,089
|
0,263
|
0,438
|
0,611
|
0,781
|
Une étude est alors menée afin de vérifier la linéarité de la méthode ainsi que ses limites de détection
et de quantification.
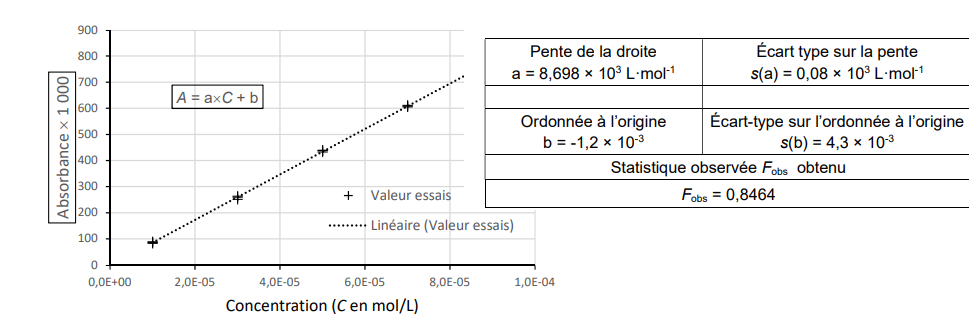 6.
6. Vérifier la linéarité de la méthode.
La courbe est une droite passant par l'origine. f
observé < f
critique.
7. Déterminer les limites de détection et de quantification. Conclure.
La limite de détection est la plus petite concentration que l'on peut distinguer du
blanc avec un risque de 0,13 %.
L
D = 3 s(b) / a =3 x4,3 10
-3 / (8,698 10
3)=1,48 10
-6 mol / L
La limite de quantification d’une
méthode est la concentration minimale qui peut être quantifiée avec un risque à 0,5 °/
00.
LD = 10 s(b) / a =10 x4,3 10-3 / (8,698 103)=4,94 10-6 mol / L.
En sortie des cuves de fermentation, après 162 heures de fermentation, le produit de synthèse est
analysé avec le spectrophotomètre UV-visible. L’absorbance mesurée dans les conditions de la
méthode décrite précédemment est de 0,387.
8. Déterminer la concentration en fluostatine C en sortie de la cuve de fermentation.
C = (A+b) / a =(0,387+1,2 10
-3) / (8,698 10
3) =4,46 10
-5 mol / L.
L’analyse par Chromatographie Liquide Haute Pression du filtrat de culture d’une souche de bactérie
Streptomyces Acta 1383 après 162 heures de fermentation révèle trois pics attribués à trois fluostatines
différentes. Par ailleurs un étalon interne a été ajouté, la caféine, afin de pouvoir quantifier les
différentes espèces présentes.
Le chromatogramme est représenté ci-dessous et les aires des différents pics sont également
fournies.
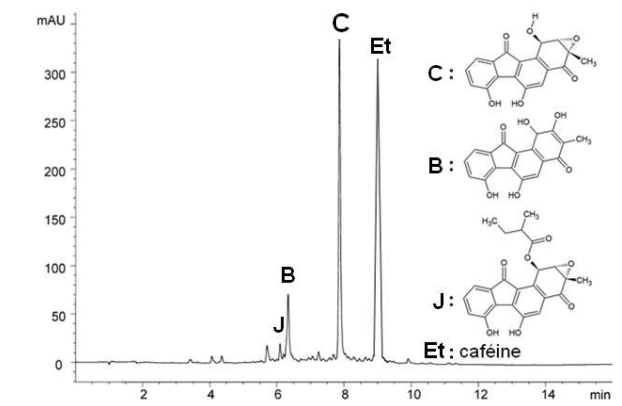
Les aires sous les pics obtenues pour l’échantillon étalon et pour le filtrat sont reportées ci-dessous.
Référence
|
Fluostatine C
|
Fluostatine J
|
Fluostatine B
|
Cafeine
|
Concentration ( mg/L)
|
5
|
5
|
5
|
10
|
Aire (mAU)
|
10,8
|
4,7
|
9,9
|
15,9
|
Filtrat
|
Fluostatine C |
Fluostatine J |
Fluostatine B |
Cafeine |
| Concentration ( mg/L) |
|
|
|
10
|
Aire (mAU)
|
24,2
|
1,0
|
4,6
|
16,4
|
9. Déterminer les concentrations en masse des trois espèces de fluostatine C, J et B contenues
dans le filtrat.
Fluostatine C : référence : aire pic caféine / aire pic fluostatine C = 15,9 /10,8 =1,47.
filtrat : aire pic caféine / aire pic fluostatine C =16,4 / 24,2 =0,678 ; 1,47 /0,678 x 5 =10,9 mg / L.
Autre méthode :
KC /ét =CC / Cét xAét / AC =5 / 10 x15,9 / 10,8 =0,736.
C'C / C'ét = KC /ét x A'C / A'ét =0,736 x 24,2 / 16,4 =1,086 ; C'C =1,086 x 10 = 10,86 ~10,9 mg / L.
Fluostatine J : référence : aire pic caféine / aire pic fluostatine J = 15,9 /4,7 =3,383.
filtrat : aire pic caféine / aire pic fluostatine J =16,4 / 1,0 =16,4 ; 3,383 /16,4 x 5 =1,03 mg / L.
Fluostatine B : référence : aire pic caféine / aire pic fluostatine B = 15,9 /9,9 =1,606.
filtrat : aire pic caféine / aire pic fluostatine B =16,4 / 4,6 =3,565 ; 1,606 /3,565 x 5 =2,25 mg / L.
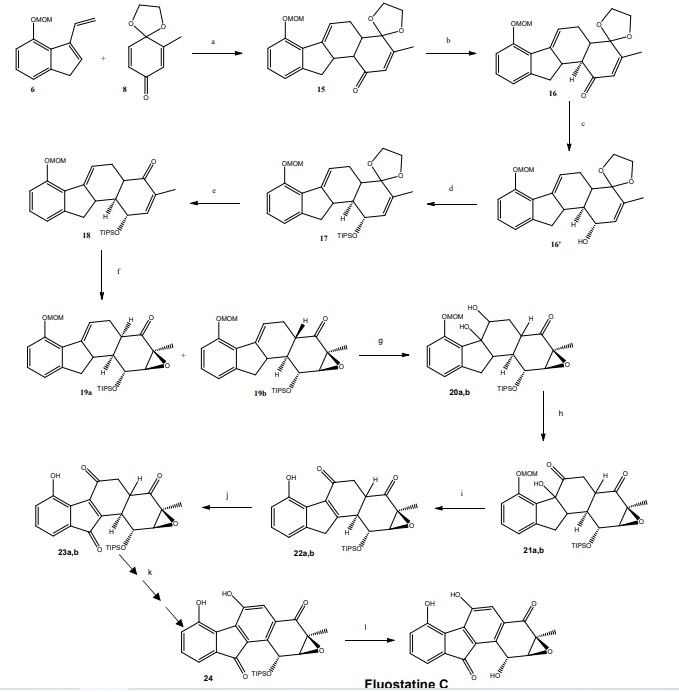
Synthèse des fluostatines C puis E.
Le travail de l’équipe de Danishefsky a pour objectif la synthèse d’un membre de cette famille qui
permettra d’obtenir d’autres fluostatines plus efficaces sur le plan thérapeutique. La fluostatine C est la
première espèce chimique de la famille obtenue. La conversion de celle-ci en fluostatine E est alors
possible.
Le composé de départ de la synthèse de la fluostatine C développée par l’équipe de Danishefsky est
le vinylindène (6), obtenu à partir de l’indanone (4).
De l’indanone (4) au vinylindène (6)
La séquence réactionnelle de cette synthèse est donnée ci-après. On rappelle que le groupement
méthoxymétyle MOM- représente le substituant H
3C-O-CH
2-.
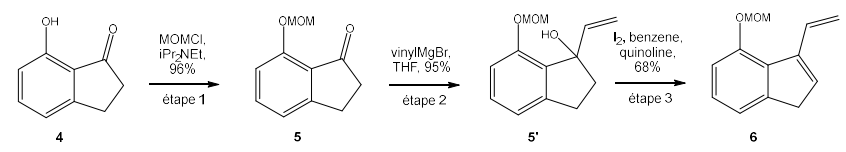 10.
10. Parmi les termes suivants : substitution, addition, élimination, préciser le nom de la réaction à
laquelle s’apparente l’étape 2.
Addition nucléophile d'un organomagnésien sur le liaison double C=O.
Le protocole expérimental de l’étape 2 est donné comme suit.
To a stirred and cooled (0 °C) solution of vinylMgBr (1.0 M, 25 mL, 25 mmol) was added dropwise a
solution of MOM protected indanone (5) (2.4 g, 12.5 mmol) in THF (10 mL). After stirring at this
temperature for 30 min, the reaction was quenched by the addition of aqueous saturated NaHCO3
(30 mL).
The mixture was extracted with EtOAc (total of 100 mL). The combined organic extracts
were washed with brine (20 mL), dried (Na2SO4) and concentrated. Flash chromatography of the
residue over silica gel (2.5 x 16 cm), using 3:1 hexane : EtOAc, gave tertiary alcohol (5’) (2.6 g, 95%)
as colorless oil.
Analysis of the colorless oil
IR spectrum (CH
2Cl
2, cast) : 3567, 2955, 1591, 1475, 1153, 1039 cm
-1
.
11. Nommer la technique utilisant l’acétate d’éthyle pur (EtOAc) dans ce protocole (indiquée en
gras). Rédiger, en quelques lignes, le protocole permettant sa mise en œuvre.
Extraction par solvant. Cette technique repose sur la solubilité de
l'espèce à extraire dans un solvant donné. Ce dernier ne doit pas être
miscible avec l'eau ; il doit dissoudre facilement l'espèce à extraire
et être liquide à la température de l'extraction.
Placer la mixture dans une ampoule à décanter et ajouter le solvant
d'extraction ; boucher et agiter ; dégazer ; placer un becher sous
l'ampoule, laisser couler le liquide le plus dense.
Les spectres RMN
13C, découplés du proton, du substrat et du produit d’intérêt obtenu lors de l’étape
2, en solution dans CDCl
3, sont donnés ci-après.
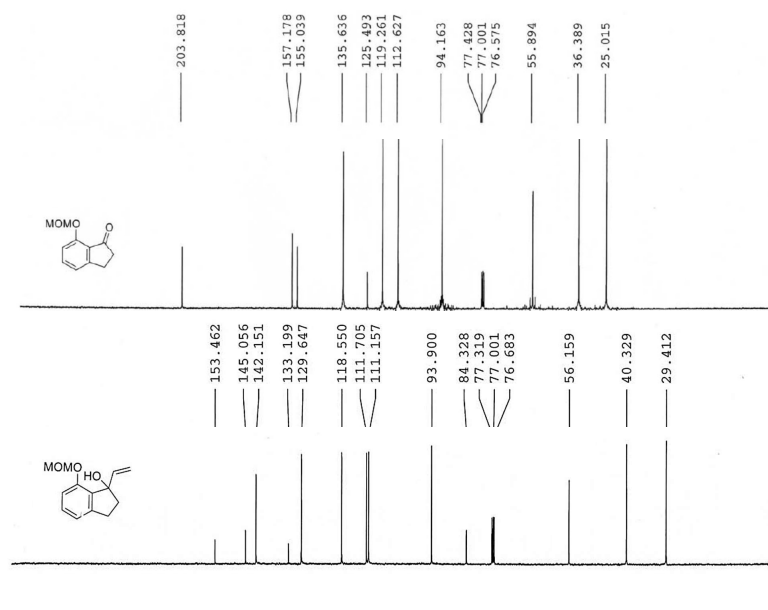 12.
12. Justifier que ces spectres permettent d’affirmer que le produit d’intérêt obtenu dans le protocole
de l’étape 2 est celui attendu et que la chromatographie flash a permis de le purifier.
Disparition du pic à 203 ppm ( carbonyle de la cétone).
Apparition des pics relatifs à la liaison double C=C vers 150 ppm.
Apparition du pic relatif à la liaison C-OH alcool vers 84 ppm.
13. Expliquer pourquoi l’étape 3 est cataloguée comme une élimination.
Elimination de H
2O : le groupe OH de l'alcol tertiaire disparaît au profit d'une liaison double C=C.
Le protocole de l’étape 3 est décrit comme suit.
A solution of tertiary alcohol (5’) (2 g, 9 mmol) in benzene (55 mL) containing I2 (50 mg) and quinoline
(0.3 mL) was heated to reflux for 2 h. After cooled to room temperature, the above solution was
concentrated to 1/3 volume, which was purified by flash chromatography over silica gel (2.5 x 18 cm),
using 4:1 hexane:ether, gave diene (6) (ca. 1.25 g, 68 %) as slightly yellowish oil, which was used
immediately in the next step.
Analysis of the yellowish oil
IR spectrum (neat) 3095, 2954, 2895, 1583, 1471, 1248, 1151 cm
-1
.
14. Justifier la nécessité de remplacer le benzène par un autre solvant. Proposer un solvant de
substitution, en justifiant le choix effectué.
Benzène : toxique, cancérogène, neurotoxique central.
Le benzène sera remplacé par le cyclohexane.
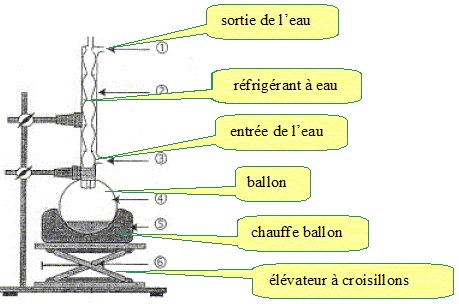
15. Représenter et légender un schéma du montage utilisé pour réaliser la synthèse du diène (6).
 16.
16. Montrer que l’analyse par spectroscopie infrarouge confirme que la transformation souhaitée a
effectivement eu lieu.
Disparition de la bande relative à O-H libre vers 3600 cm
-1.
Apparition d'une bande vers 1583 cm
-1, double liaison C=C conjuguée.
Chromatographie flash.
L’équipe a choisi, dans cette synthèse totale et pour chaque étape, une technique de purification
appelée chromatographie flash, applicable aussi bien aux solides qu’aux liquides.
17. Citer les deux techniques courantes de purification utilisées au laboratoire, remplacées ici par
la chromatographie flash.
Filtration, centrifugation, évaporation, extraction.
18. Proposer une technique d’analyse courante, au laboratoire, se rapprochant de la
chromatographie flash.
Chromatographie sur colonne.